
Research
Genome-wide molecular dating
Zuckerkandl and Pauling showed 50 years ago that molecular sequences are ‘documents of evolutionary history’. The composition of the molecular sequences of organisms living today has been passed on from ancestors from long ago. Unused segments get lost, new parts are added and small changes occur because replication is not error free. Whatever modifications have happened, they will have left traces in the molecular sequences.
In the advent of next generation sequencing technologies we have thousands of genomes available, however, we lack methods for genome-wide molecular dating. In this project, we will develop novel methods which will model not only mutations but also processes such as gene duplication, loss and transfer. We will also consider variation within species and devise methods that allow to use the genome-wide DNA sequences of multiple individuals per species to perform molecular dating. Link to project website https://synergy.st-andrews.ac.uk/genomemoleculardating/
Polymorphism-Aware Phylogenetic Models
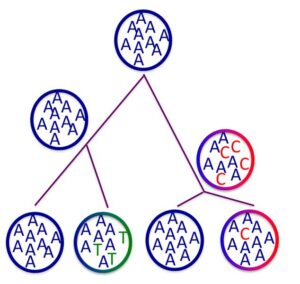
We have developed a new method called Polymorphism-aware phylogenetic model (PoMo). PoMo is a phylogenetic Markov model with states that represent fixed alleles and states representing polymorphic alleles at different frequencies. A substitution is thus modeled from mutation, through the transient polymorphic stage, and to fixation (or loss). Polymorphic states can be observed in the tips of the phylogeny as well as at ancestral nodes. We have developed applications of this novel approach to the inference of evolutionary parameters, such as mutation and selection, and to the estimation of phylogenies. PoMos are alternative approaches of species tree estimation that add a new layer of complexity to the standard substitution models by accounting for population-level forces to describe the process of sequence evolution. Overall, PoMos constitute a full-likelihood yet computationally efficient approach to species tree inference. PoMos are designed to cope with recent radiations, including incomplete lineage sorting, and long divergence times. PoMos have recently been implemented as part of the RevBayes statistical platform for Bayesian phylogenetic inference.
Finding targets of selection in experimental evolution
We developed a Bayesian inference method designed for Evolve & Resequence (E&R) – Bait-ER – that infers selection coefficients and tests for selection. It analyses full allele frequency trajectories while looking for consistency across experimental replicate populations. Bait-ER also accounts for added noise due to pooled sequencing.